Background
Kidney cysts can be found in several conditions affecting children. These cysts can result from nonhereditary fetal abnormalities or genetic conditions; in rare cases, they may develop later in life.
Polycystic kidney disease, a disorder that can be diagnosed in adult and pediatric patients, is an inherited disease that involves bilateral renal cysts without dysplasia. The condition is broadly divided into two forms:
-
Autosomal recessive polycystic kidney disease (ARPKD)
-
Autosomal dominant polycystic kidney disease (ADPKD)
ARPKD, previously known as infantile polycystic kidney disease, occurs in about 1 in 20,000 live births, with a carrier rate of 1 in 70 individuals. The age at which symptoms appear can vary: roughly one third of patients show signs before the age of 1 year, another third between the ages of 1 and 20 years, and the remaining third after 20 years of age.
ADPKD, previously known as adult polycystic kidney disease, is a prevalent genetic condition, occurring in approximately 1 in 400 to 1 in 1000 live births. Individuals with ADPKD typically do not experience symptoms until adulthood. Often, ADPKD is detected in asymptomatic children during abdominal imaging, either as part of a screening owing to a family history of ADPKD or incidentally for an unrelated concern. However, a minority of patients with ADPKD present during infancy or childhood with an early onset and rapid progression. See the image below.
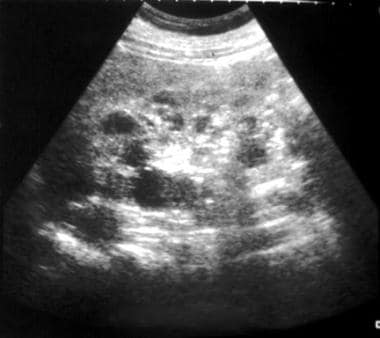 Sonogram shows cysts with bilaterally enlarged kidneys. These findings are compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
Sonogram shows cysts with bilaterally enlarged kidneys. These findings are compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
The nomenclature of infantile versus adult is no longer used because it is not an accurate description. ARPKD and ADPKD can involve the presence of renal cysts at any time during an affected person's life, from the prenatal period to adolescence or older. The clinical and radiologic manifestations of both types of polycystic kidney disease have considerable overlap.
Autosomal recessive polycystic kidney disease
ARPKD is characterized by cystic dilatation of renal collecting ducts associated with hepatic abnormalities of varying degrees, including biliary dysgenesis and periportal fibrosis. ARPKD was first recognized in 1902; however, the histology was not reported until 1947. In 1964, Osathanondh and Potter classified ARPKD as type 1 cystic kidney disease. [1, 2] Eventually, because neither parent had the disease and no sex predilection was observed, this disease was concluded to have an autosomal recessive mode of inheritance. See the image below.
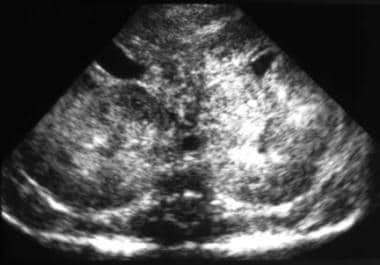 Sonogram shows enlargement of both kidneys, diffuse increased echogenicity, and loss of corticomedullary differentiation. These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
Sonogram shows enlargement of both kidneys, diffuse increased echogenicity, and loss of corticomedullary differentiation. These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
ARPKD was originally described as four separate clinical entities based on age of presentation. This classification is no longer considered valid because of the large degree of overlap among the different groups and the wide range of possible presentations, regardless of age. ARPKD is primarily caused by variants in the PKHD1 gene, located on chromosome 6p21. This gene encodes fibrocystin (also known as polyductin), a large integral membrane protein. To date, over 750 PKHD1 variants have been identified. Most individuals with ARPKD are compound heterozygotes, meaning they inherit two different mutant alleles.
Fibrocystin is present in the kidney's collecting ducts and thick ascending limb, as well as in the epithelial cells of the hepatic bile duct. It is also found in the liver, pancreas, and lungs. Although fibrocystin is co-expressed with proteins linked to ADPKD, it does not share structural similarities with these proteins. This indicates that abnormalities in fibrocystin may impair the proper functioning of renal cilia, suggesting a common mechanism underlying cyst formation in both conditions.
Autosomal dominant polycystic kidney disease
ADPKD is the most common inherited kidney disease in humans. It is a multisystem disorder characterized by progressive cystic dilatation of both kidneys (see the image below), with variable extrarenal manifestations in the gastrointestinal (GI) tract, cardiovascular system, reproductive organs, and brain. [3]
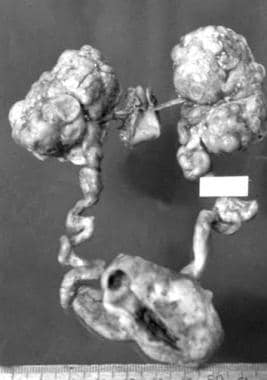 Pathologic specimen of end-stage autosomal dominant polycystic kidney disease (ADPKD) with deformed lobulated kidneys.
Pathologic specimen of end-stage autosomal dominant polycystic kidney disease (ADPKD) with deformed lobulated kidneys.
Hepatic cysts are possible in ADPKD, although they are less common than in ARPKD. ADPKD has a wide clinical spectrum.
Etiology
The three basic processes involved in renal cyst formation and progressive enlargement are as follows [4, 5] :
-
Tubular cell hyperplasia
-
Tubular fluid secretion
-
Abnormalities in tubular extracellular matrix and/or function
Tubular cell hyperplasia
This may be mediated by factors that control cell proliferation (eg, epidermal growth factor, transforming growth factor-α), dysregulation of apoptosis, or the balance between the two.
Tubular fluid secretion
The solid tumor cell nests produced by the cell hyperplasia described above are transformed into fluid-filled cysts by the secretion of fluid by the tubular cells associated with efferent tubular obstruction or slow or absent afferent flow. This accounts for the fluid within the cysts of kidneys in patients with ADPKD, 70% of which have no afferent or efferent tubular connections.
Abnormalities in tubular extracellular matrix and/or function
These are thought to be responsible for amplifying tubular cell hyperplasia and tubular fluid secretion. Interstitial inflammation and fibrosis are responsible for progression in all forms of polycystic kidney disease.
Autosomal recessive polycystic kidney disease
In 1994, the ARPKD gene (PKDHD1) was localized to the short arm of chromosome 6. [6] Fibrocystin/polyductin, a protein encoded by PKDHD1, is expressed on the cilia of renal and bile duct epithelial cells and is thought to be crucial in maintaining the normal tubular architecture of renal tubules and bile ducts. However, the precise function of this protein has yet to be completely studied or understood. The protein strengthens the theory that the primary defect in ARPKD is linked to ciliary dysfunction. [7]
ARPKD is characterized by nonobstructive, bilateral, symmetrical dilatation and elongation of 10-90% of the renal collecting ducts, focally accounting for a wide variability of renal dysfunction. As the number of ducts involved increases, the kidneys enlarge. However, at autopsy, the reniform shape is maintained, because the abnormality is in the collecting ducts and the cysts are usually minute (< 3 mm). In older patients, cysts as large as 10 mm may be seen. (See the images below.)
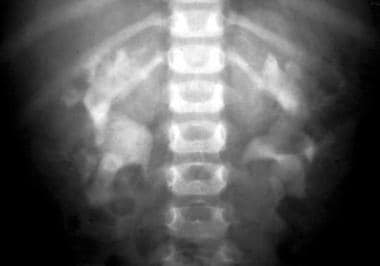 Excretory urogram shows minimal bilateral tubular changes caused by a mild form of autosomal recessive polycystic kidney disease (ARPKD).
Excretory urogram shows minimal bilateral tubular changes caused by a mild form of autosomal recessive polycystic kidney disease (ARPKD).
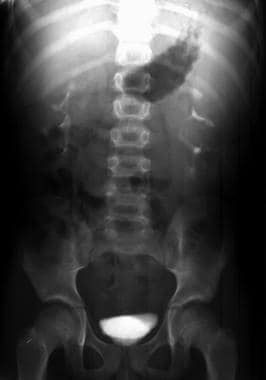 Excretory urogram shows enlarged kidneys with bilateral distortion of the collecting system (spider-legs configuration). These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
Excretory urogram shows enlarged kidneys with bilateral distortion of the collecting system (spider-legs configuration). These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
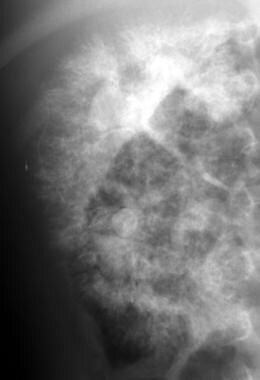 Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
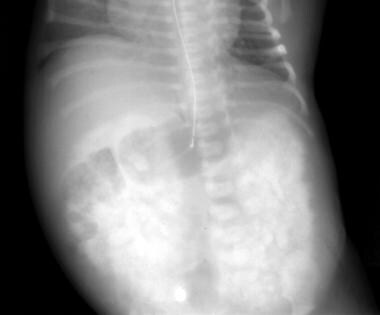 Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
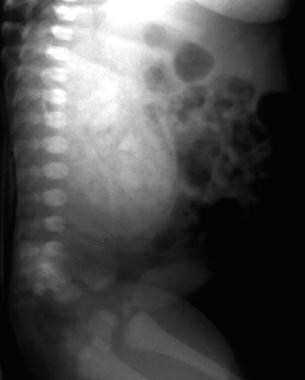 Excretory urogram shows the typical mottled (spongelike) contrast enhancement pattern in autosomal recessive polycystic kidney disease (ARPKD).
Excretory urogram shows the typical mottled (spongelike) contrast enhancement pattern in autosomal recessive polycystic kidney disease (ARPKD).
At autopsy, gross examination of a kidney in patients with ARPKD reveals multiple minute cystic spaces throughout the capsular surfaces. Cut sections of the kidney show that these cystic structures are subcapsular extensions of radially oriented cylindrical or fusiform ectatic spaces, with poor corticomedullary differentiation due to the extension of the elongated and dilated collecting ducts from the medulla to the cortex.
All patients with ARPKD have congenital hepatic fibrosis (CHF), which may have a more severe clinical manifestation than the renal disease. The CHF results from malformation of the developing ductal plate. The liver biopsy findings reveal enlarged, fibrotic portal tracts and hyperplastic, dilated, and dysgenetic biliary ducts with normal hepatocytes. The ductules can show true cystic changes, and, when the changes are macroscopic, ARPKD can be indistinguishable from Caroli disease. The portal hypertension secondary to the CHF can be clinically debilitating, with splenomegaly, varices, and GI hemorrhage. [8]
The results from one study noted that characteristics of CHF are similar in both autosomal dominant and autosomal recessive polycystic kidney diseases. [9]
In a study designed to better understand the complications of ARPKD, researchers at the National Institutes of Health (NIH) analyzed clinical, molecular, and imaging data from 73 patients (aged 1-56 years; average, 12.7 years) with mutations in PKHD1 and kidney and liver involvement. The findings identified platelet count as the best predictor of the severity of portal hypertension, which has early onset but is underdiagnosed in patients with autosomal recessive polycystic kidney disease. [10]
A study that included 304 patients with ARPKD found that biallelic null variants in PKHD1 are associated with severe disease and a poor prognosis. For example, variants that affect amino acids 2625-4074 of fibrocystin are linked to worse hepatic outcomes. [11]
Autosomal dominant polycystic kidney disease
The genes responsible for ADPKD were localized to the short arm of chromosome 16 (PKD1) in 85% of cases and the long arm of chromosome 4 (PKD2) in most of the remaining cases. The proteins encoded by PKD1 and PKD2 are polycystin 1 and polycystin 2, respectively. These proteins are expressed in the developing kidney, and their functions overlap considerably.
The dysfunction of these proteins is thought to be pathogenetically responsible for the manifestations of ADPKD, primarily by renal ciliary dysfunction. Whether a third gene accounts for a small number of unlinked families is uncertain. Homozygous or compound heterozygous genotypes have been thought to be lethal in utero. Individuals heterozygous for PKD1 and PKD2 mutations usually survive to adulthood but have more severe renal disease.
ADPKD differs from ARPKD in that cysts associated with ADPKD develop anywhere along the nephron. Upon clinical presentation, kidneys are usually enlarged, with numerous large, round nodules on the external surface of the kidney, causing the loss of its original reniform shape, which is different from kidneys in patients with ARPKD.
Cysts of varying sizes containing pale fluid or blood are randomly distributed throughout the parenchyma and involve any segment along the nephron. The cysts have thickened basement membranes with pericystic interstitial fibrosis, and their epithelium maintains active secretion and reabsorption. It has been hypothesized that patients with an associated marked epithelial hyperplasia may have a higher rate of malignant transformation than does the general population.
Epidemiology
United States statistics
Autosomal recessive polycystic kidney disease
The exact incidence of ARPKD is unknown because of varying reports in patient autopsies versus survivors, as well as the possibility of affected children who die perinatally without a definitive diagnosis. The frequency of ARPKD has been reported as 1 case per 10,000-40,000 births, although the frequency of the gene in the general population is estimated to be 1 case per 70 population.
Because of the recessive inheritance of ARPKD, both parents are unaffected. The recurrence risk in subsequent pregnancies is 25%. Unaffected siblings have a 66% chance of being carriers. Carriers or heterozygotes are asymptomatic.
Autosomal dominant polycystic kidney disease
The estimated prevalence of ADPKD is 1 case per 200-1000 population. ADPKD is responsible for 6-10% of cases of end-stage renal disease in North America. Because of the autosomal dominant inheritance, one parent is usually affected, and each offspring has a 50% chance of inheriting the gene, with a penetrance of almost 100%.
International statistics
ADPKD is responsible for 6-10% of cases of end-stage renal disease in Europe.
Race- and sex-related demographics
Both forms of polycystic kidney disease affect all racial and ethnic groups, and both equally affect males and females.
Prognosis
Determining the prognosis of polycystic kidney disease is difficult; however, with advances in medical management and continued progress in end-stage renal disease therapy in young infants, further improvements in survival and rehabilitation can be expected.
Autosomal recessive polycystic kidney disease
The clinical manifestations of ARPKD vary depending on the number of collecting ducts involved, as well as the degree of interstitial fibrosis. Fetuses with severe impairment of renal function and reduced fetal urinary output present with oligohydramnios, which may result in pulmonary hypoplasia. Most of these infants die from pulmonary complications after birth. [12]
Babies with less severe renal manifestations who survive the neonatal period may still develop chronic kidney disease, which occurs at varying ages depending on the degree of renal involvement. Pulmonary insufficiency with respiratory distress due to oligohydramnios that is worsened by large renal masses is a major cause of morbidity and mortality in neonates.
In patients who survive the neonatal period, renal prognosis has improved over time because of renal transplantation. CHF still causes considerable morbidity, even in patients who have received transplants; some die from GI hemorrhage secondary to portal hypertension. Oliguric acute renal failure (ARF) often improves as the pulmonary function improves.
Autosomal dominant polycystic kidney disease
ADPKD can also present prenatally but usually does not involve the severe renal impairment seen in ARPKD. In adults, it more commonly causes chronic kidney disease that progresses to further cystic development of the renal cortex, often with transition into end-stage renal disease. Thus, the chance of end-stage renal disease is 2% in patients younger than age 40 years and increases to 50% by the seventh decade of life.
ADPKD is a multisystem disorder, and some patients develop associated intracranial aneurysms, which can cause stroke and intracranial hemorrhage. Much of the morbidity of ADPKD is due to chronic hypertension. A study by Seeman et al showed that the prevalence of hypertension rose from 20% to 38%, along with an increase in the number of kidney cysts, during a median 6-year follow-up period in children. [13] ADPKD can manifest in utero with the Potter phenotype, with death from pulmonary hypoplasia. [14]
Patient Education
The Polycystic Kidney Disease (PKD) Foundation is devoted to determining the cause of polycystic kidney disease, improving its clinical treatment, and discovering a cure. To become members, patients, family members, friends, physicians, and allied health professionals can contact the foundation at the following:
1001 E. 101st Terrace, Suite 220
Kansas City, MO 64131
Local phone: 816.931.2600
Email: pkdcure@pkdcure.org
Additional information can be obtained by contacting the National Kidney Foundation at the following:
National Kidney Foundation
30 East 33rd Street
New York, NY 10016
-
Sonogram shows cysts with bilaterally enlarged kidneys. These findings are compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
-
Sonogram shows cysts with bilaterally enlarged kidneys. These findings are compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
-
Sonogram shows cysts with bilaterally enlarged kidneys. These findings are compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
-
Frontal excretory urogram of autosomal dominant polycystic kidney disease (ADPKD) shows a spider-legs configuration of the collecting system secondary to compression due to cysts.
-
Lateral excretory urogram of autosomal dominant polycystic kidney disease (ADPKD) shows a spider-legs configuration of the collecting system secondary to compression due to cysts.
-
Pathologic specimen of end-stage autosomal dominant polycystic kidney disease (ADPKD) with deformed lobulated kidneys.
-
Sonogram shows enlargement of both kidneys, diffuse increased echogenicity, and loss of corticomedullary differentiation. These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
-
Excretory urogram shows minimal bilateral tubular changes caused by a mild form of autosomal recessive polycystic kidney disease (ARPKD).
-
Excretory urogram shows enlarged kidneys with bilateral distortion of the collecting system (spider-legs configuration). These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
-
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
-
Excretory urogram shows the typical mottled (spongelike) contrast pattern in autosomal recessive polycystic kidney disease (ARPKD).
-
Excretory urogram shows the typical mottled (spongelike) contrast enhancement pattern in autosomal recessive polycystic kidney disease (ARPKD).
-
CT shows bilaterally smooth enlarged kidneys. These findings are compatible with a diagnosis of autosomal recessive polycystic kidney disease (ARPKD).
-
CT shows bilateral renal and liver cysts with enlarged kidneys and remaining renal cortex enhancement compatible with a diagnosis of autosomal dominant polycystic kidney disease (ADPKD).
-
T2-weighted MRI shows bilateral smooth enlarged kidneys with a hyperintense, linear, radial pattern in the cortex and medulla, compatible with autosomal recessive kidney disease.
-
T1- and T2-weighted MRIs demonstrating a superior left kidney cyst with high T1 and intermediary T2 signal compatible with a bleeding cyst in autosomal dominant polycystic kidney disease (ADPKD).
-
T1- and T2-weighted MRIs demonstrating bilateral renal and liver cysts compatible with autosomal dominant polycystic kidney disease (ADPKD).
